Immediately in Effect (IIE) SOP Premarket Data Issues
Immediately in Effect (IIE) SOP Premarket Data Issues
Purpose
This Standard Operating Procedure (SOP) describes the process that the Center for Devices and Radiological Health (CDRH) must follow to clarify and more quickly inform stakeholders when CDRH changes its expectations about
- new scientific information that could affect data submitted as part of an Investigational Device Exemption (IDE), or
- premarket submission.
What does premarket submission include?
Premarket submission includes the following:
- Premarket notification (510(k))
- Premarket Approval (PMA), or
- Humanitarian Device Exemption (HDE), that includes combination products that
- contain a device constituent part for which CDRH has jurisdiction, and
- need to be disseminated in a timely manner.
Public health emergency
On February 4, 2020, the Secretary of Health and Human Services determined that a public health emergency exists nationwide as a result of confirmed cases of COVID-19. During this public health emergency, FDA may deviate from procedures outlined in this SOP, for Level 1 Immediately in Effect Guidance Documents addressing this public health emergency.
Reference: For more information, contact [email protected].
Contents
The table below lists the contents of this SOP.
| Topic |
|---|
| 1. Implementation Guidelines and Background |
| 2. Premarket IIE Document Procedures |
| 3. References |
| 4. Appendices |
1.Implementation Guidelines and Background
Contents
The table below lists the topics in this section.
1.1.Implementation Guidelines
1.1.Implementation Guidelines
General scenario
For implementation in a general scenario, consider the issues raised by new scientific information with the benefit of input from other stakeholders who have expertise and viewpoints.
Even though these outside-the agency viewpoints may differ, they may add appropriate balance.
Instance of immediate implementation
Immediate implementation is applicable in the following scenarios:
- Public health emergencies
- When new premarket regulatory expectations outweigh the need for pre-implementation feedback from affected stakeholders
Mode of immediate implementation
During instances of immediate implementation, CDRH does the following:
Step |
Action |
|---|---|
1 |
Communicate through Level 1, Immediately in Effect (IIE) Guidance Documents using Good Guidance Practices (GGPs). |
2 |
Publish a Federal Register document announcing the guidance, post the IIE guidance documents on its website. |
3 |
Contemporaneously issue other relevant communication to the affected industry stakeholders summarizing the information in the IIE guidance, if necessary. |
Note: FDA’s guidance documents do not
- create or confer any right for or on any person, and
- operate to bind FDA or the public.
1.2.Background
Change implementation - current trend
Currently, manufacturers learn of the changes soon after or at the time the CDRH implements and submits them.
The changes typically are regarding data or how to gather specific data in support of an IDE, 510(k), PMA, or HDE.
Change implementation - review
Reviewers may implement changes on a case-to-case basis with immediate supervisory concurrence when it is necessary to protect public health. The changes entail
- requesting new clinical data, or
- using a new test method.
Example
A reviewer may request that sponsors test their implantable device for durability because new data demonstrates that this type of device is prone to failure due to premature wear and tear of the technology.
Publishing time
Although CDRH may issue a detailed guidance document, the document may not publish until a year or more after a Branch- or a Division-level decision has been made to request the information because of the resource constraints in developing guidance documents.
Solution to the delay
CDRH believes that timely communication with industry about changes in premarket regulatory expectations is important.
- FDA’s GGP regulation: FDA’s GGP regulation provides a mechanism for communicating and implementing certain changes in regulatory expectations quickly, without requiring prior public comment.
- FDA’s Immediately in Effect (IIE) guidance: Under 21 CFR 10.115(g)(2), FDA may issue an IIE guidance when prior public participation is not “feasible or appropriate.”
Preamble: final GGP’s rule
FDA identified the following three examples in which IIE guidance could be appropriate because prior public comment may not be feasible or appropriate:
- Public health reasons for the immediate implementation of the guidance document
- Statutory requirement, executive order, or court order that requires immediate implementation
- The guidance document that presents a less burdensome policy that is consistent with public health
Guidelines towards IIE: CDRH
CDRH intends to follow the guidelines below toward IIE:
- Use the procedures described in 21 CFR 10.115(g)(2) to issue guidance documents addressing changes in premarket regulatory expectations.
- Open a public docket upon issuance of the guidance through a Notice of Availability (NoA) in the Federal Register, and, subsequently, make changes to the guidance, if appropriate, based on public comment.
- Contemporaneously issue letters to affected industry stakeholders summarizing the information in the IIE guidance under specific circumstances.
- Post the IIE guidance documents on the FDA website.
To cater to this, CDRH has developed this SOP to facilitate issuance of such guidance documents.
Note: This SOP is being implemented after considering the comments on this topic.
2.Premarket IIE Document Processes
Contents
The table below lists the topics in this section.
2.1.Developing the Premarket IIE Guidance Document: Prerequisites and Process
2.1.Developing the Premarket IIE Guidance Document: Prerequisites and Process
Prerequisites to initiate development
To initiate the development of a premarket IIE guidance document, ensure the following prerequisites are met:
New scientific risks
New scientific information (e. g., product complaints/ recalls or unique risks that were not previously identified such as a new failure mode identified during clinical use of the product) has been identified that
- raises a new, important safety risk, or
- demonstrates that currently used test methods or clinical trial designs are inadequate to demonstrate safety, effectiveness, and/or substantial equivalence of a device type.
Changed regulatory methods
As a result of the new scientific risks that have come to light, CDRH has changed its regulatory expectations, such as a change in the data that would be expected to be provided as part of an IDE, PMA, 510(k), or HDE because it is necessary to support an IDE approval or premarket approval or clearance.
Risk to public health outweighs pre-implementation feedback
Consistent with 21 CFR 10.115(g)(2), prior public participation is not appropriate or feasible because the immediate risk to public health outweighs the need for pre-implementation feedback from affected stakeholders.
Initiating development – Process
The table below describes the stages in initiating development of a Premarket IIE Guidance:
Note: Ensure the prerequisites are met before initiating the development.
Reference: For more information about prerequisites about initiating the development, refer to Prerequisites to initiate development .
Stage |
Who |
Does What |
|---|---|---|
1 |
|
|
2 |
Staff |
Confirm that other CDRH Offices or FDA Centers are not implicated in any change resulting from the new scientific information. |
3 |
|
Concurs to follow the process outlined in Stages in Developing the Premarket IIE Guidance document instead of CDRH’s standard guidance development process. |
Developing the Premarket IIE Guidance document – Process
The table below describes the stages of development of the Premarket IIE Guidance Document:
Stage |
Who |
Does What |
|---|---|---|
1 |
Appropriate staff and management |
Present a briefing summary to the Center Science Council (CSC). |
2 |
CSC |
|
3 |
Senior CDRH leadership |
Reviews CSC’s recommendations to see if it meets regulatory and legal standards, such as the regulatory standard for issuance of IIE guidance. |
4 |
Appropriate staff and management |
|
5 |
|
|
6 |
Center Director |
Provides final clearance of the Premarket IIE Guidance. |
7 |
Appropriate staff |
|
8 |
Appropriate staff |
2.2.Presenting the Summary to the Center Science Council
-
Summary presentationRead Block
-
Determining the responseRead Block
-
CSC’s response on changes to premarket regulatory expectationsRead Block
-
CSC’s communication about change in regulatory expectationsRead Block
-
CSC’s response to topics needing additional expertiseRead Block
-
Reviewing CSC’s recommendationRead Block
2.2.Presenting the Summary to the Center Science Council
Summary presentation
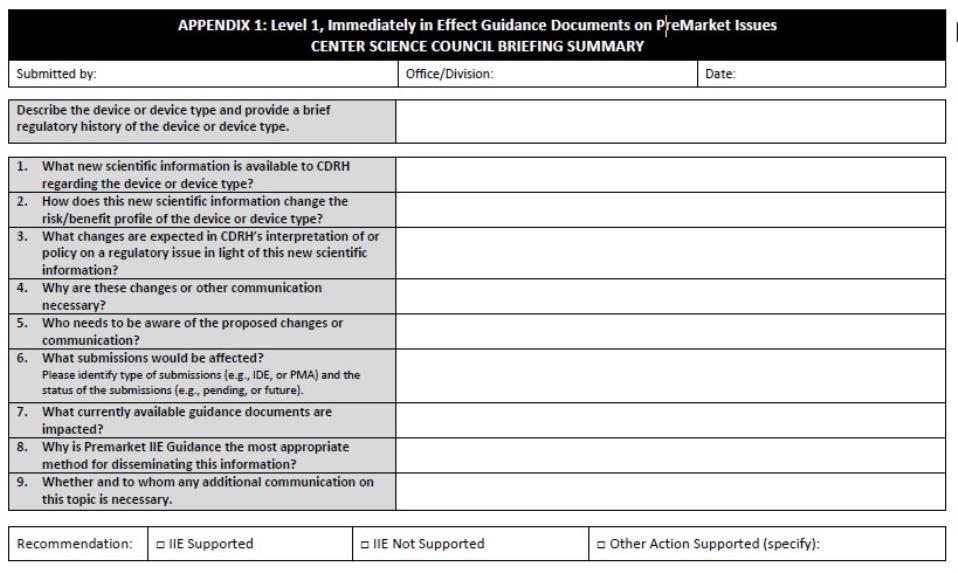
The appropriate staff and their management must present the Center Science Council (CSC) with a briefing summary of the following:
Reference: See Appendix 1 for the Premarket IIE Guidance Document Center Science Council Briefing Summary.
- The new scientific information available to CDRH
- How this new scientific information changes the risk/ benefit profile of the device or device type
- Any expected change in the Center’s interpretation of or policy on a regulatory issue in light of this new scientific information
- Why such proposed changes are necessary
- The audience who should be aware of such proposed changes or communication
- What submissions should be affected (e.g., pending/future premarket submissions, pending/future IDE submissions)
- What existing guidance documents are impacted, if any
- Why a Premarket IIE Guidance Document is the only appropriate method for disseminating this information, and
- Whether and to whom any additional communication on this topic is necessary.
Determining the response
Based on the information and discussion points presented to CSC, the CSC determines the following for an appropriate response:
CSC’s response on changes to premarket regulatory expectations
The table below describes CSC’s response on receiving new information warranting change in premarket regulatory expectations.
If the new information ... |
Then the CSC must... |
|---|---|
warrants a change in premarket regulatory expectations |
communicate using
|
does not warrant a change in the regulatory expectations |
determine whether it must issue a separate communication alerting stakeholders to new scientific information. Note: Such communication is not addressed in this SOP. |
CSC’s communication about change in regulatory expectations
If the Center is proposing a change in the regulatory expectation, then the CSC may provide a recommendation whether
- a traditional guidance document should be issued in draft and finalized, or
- the Center should issue a Premarket IIE Guidance Document under this SOP owing to any of the following reasons:
- There are public health reasons for the immediate implementation of the guidance document.
- There is a statutory requirement, executive order or court order that requires immediate implement.
- The guidance document presents a less burdensome policy that is consistent with public health.
CSC’s response to topics needing additional expertise
If the new information needs additional expertise for an appropriate response, the CSC follows appropriate mechanisms to obtain external expertise.
Reviewing CSC’s recommendation
The Senior CDRH leadership reviews CSC’s recommendation to ensure that CSC’s recommendations meet regulatory and legal standards, such as the regulatory standard for issuance of IIE guidance.
2.3.Drafting the Premarket IIE Guidance Document
2.3.Drafting the Premarket IIE Guidance Document
Stages in drafting the IIE Guidance Document
The table below describes the developmental stages of the IIE Guidance Document:
Stage |
Description |
|---|---|
1 |
|
2 |
|
3 |
IIE Guidance template
Use Appendix 2 as a template to develop the Premarket IIE Guidance document.
Reference: For more information about the template, refer to Appendix 2: IIE Guidance Documents on Premarket Issues .
Note: The Premarket IIE Guidance document must ideally be 1-2 pages, and generally no more than 5 pages in length.
Developing the Premarket IIE Guidance content
While developing the Premarket IIE Guidance document, do the following:
Step |
Action |
||||||
|---|---|---|---|---|---|---|---|
1 |
Identify the following:
|
||||||
2 |
Discuss why this new scientific information warrants a change in regulatory expectations for the device type or is of importance to the specified audience. |
||||||
3 |
Explain why public comment prior to issuance is not feasible or appropriate such that a Premarket IIE Guidance document is being issued. |
||||||
4 |
Outline the changes in regulatory expectations. For example, such as changes in data expected to be submitted in light of this new scientific information. |
||||||
5 |
|
||||||
6 |
Identify and include the following:
|
Additional reference
Reference any other communication or publicly available information from CDRH related to the issue discussed in the Premarket IIE Guidance.
2.4.Reviewing and Clearing of the Premarket IIE Guidance Document
Stages in review and clearance
The table below describes the stages of review and clearance of the Premarket IIE Guidance:
Stage |
Who |
Does What |
|---|---|---|
1 |
GGP Representative |
|
2 |
|
Reviews and clears the Premarket IIE Guidance with respect to content and Office-level policy. |
3 |
CDRH Deputy Directors for Policy and Science |
Review and clear the Premarket IIE Guidance with respect to content and Center-level policy. |
4 |
Office of Chief Counsel (OCC) |
Reviews the Premarket IIE Guidance for legal sufficiency and accuracy. |
5 |
Center Director |
Provides a final clearance of the Premarket IIE Guidance. |
2.5.Issuing the Premarket IIE Guidance Document
2.5.Issuing the Premarket IIE Guidance Document
Guidelines
Adhere to the following guidelines while issuing the Premarket IIE Guidance document:
- Post all Premarket IIE Guidance in one readily accessible location.
- Determine any other appropriate methods for distribution, such as email or postal mail.
- Identify the relevant stakeholders to distribute additional communication on this topic as appropriate.
- Determine the appropriate mechanism for distribution of additional communication.
Issuing the document
Issue premarket IIE Guidance as follows:
Step |
Action |
|---|---|
1 |
Publish an NoA in the Federal Register announcing availability of the Premarket IIE Guidance and requesting comments. |
2 |
Post a copy of the Premarket IIE Guidance on the CDRH website. |
2.6.Reviewing Comments and Revising the Premarket IIE Guidance
Reviewing by staff
The table below describes how to determine if the document needs to be revised based on the comments:
Step |
Action |
||||||
|---|---|---|---|---|---|---|---|
1 |
Collect comments from the docket within 60 days after the Premarket IIE Guidance was published. |
||||||
2 |
|
||||||
3 |
Maintain an administrative record of the comments and revisions. |
||||||
4 |
Present the recommendations to the CSC for discussion.
Note: Publicly report the number of Premarket IIE Guidance issued at periodic intervals. |
3.References
Contents
3.1.Abbreviations
List of terms and their full forms
The table below provides the full forms of the abbreviated terms used within this SOP.
Term |
Full Form |
|---|---|
CDRH |
Center for Devices and Radiological Health |
CFR |
To be added |
CSC |
Center Science Council |
GGP |
Good Guidance Practice |
HDE |
Humanitarian Device Exemption |
HFA |
To be added |
IDE |
Investigational Device Exemption |
IIE |
Immediately in Effect |
NoA |
Notice of Availability |
OCC |
Office of Chief Counsel |
ODE |
Office of Device Evaluation |
OIR |
Office of In Vitro Diagnostics and Radiological Health |
PMA |
Premarket Approval |
SOP |
Standard Operating Procedure |
3.2.Change History
Change Control Table
Following is the change control table listing effective dates pertaining to this SOP:
Version Number |
Reason for Change |
Effective Date |
Approving Official (Name/Title) |
|---|---|---|---|
1.0 |
Original |
Draft issued September 5, 2013 |
Jeffrey Shuren, MD, JD Director, Center for Devices and Radiological Health |
2.0 |
Finalizing Draft based on comments received |
March 26, 2014 |
Jeffrey Shuren, MD, JD Director, Center for Devices and Radiological Health |
4.Appendices
Contents
The table below lists the topic(s) in this chapter.
| Topic |
|---|
| 4.1. Appendix 1: Level 1, Immediately in Effect Guidance Documents on Premarket Issues |
| 4.2. Appendix 2: IIE Guidance Documents on Premarket Issues |